It looks like nothing was found at this location. Maybe try a search?
Click here to listen to Uveitis and Steroid-Sparing Therapy
Presented by C. Stephen Foster, MD, FACS, FACR
Audio-Digest Ophthalmology Volume 56, Issue 15
Interactive Blog Summary
Submit a Question
Click on the Register link
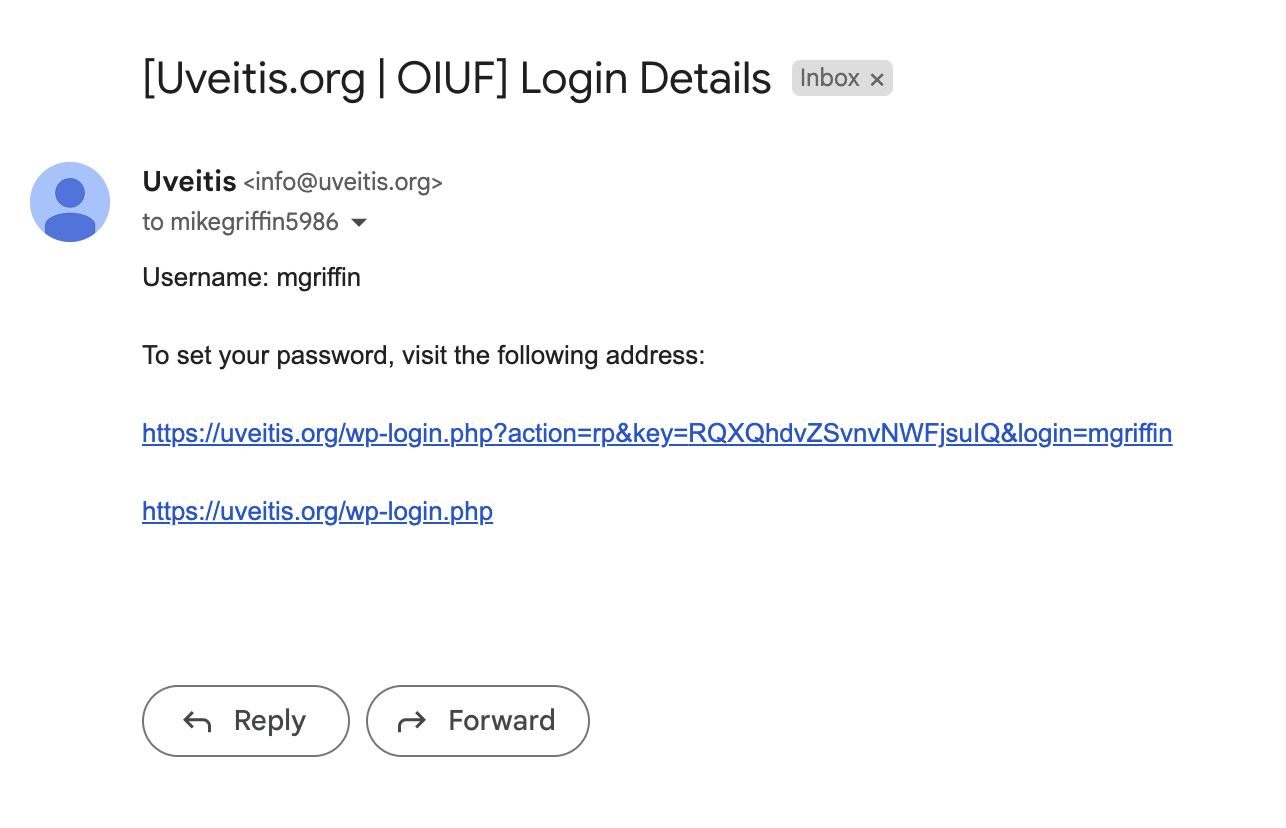
In the email you receive, click on the change password link.
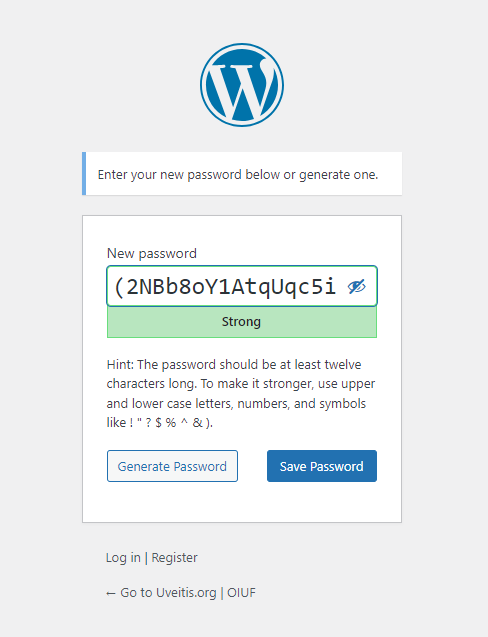
Change your password