Juvenile Idiopathic Arthritis and Uveitis
C. Stephen Foster, M.D., F.A.C.S., F.A.C.R.
Uveitis is a serious complication of juvenile idiopathic arthritis (JIA). Approximately 6% of all cases of uveitis occur in children, and up to 80% of all cases of anterior uveitis in childhood are associated with JIA. Although remarkable progress has been made in the care of patients with JIA-associated uveitis since the development of corticosteroids for systemic and ophthalmic use in the 1950’s, up to 12% of children with uveitis associated with pauciarticular JIA still develop permanent blindness as a result of low-grade chronic intraocular inflammation. Ironically, these children are often under careful observation by ophthalmologists who may opt to tolerate low-grade ocular inflammation, hoping to avoid the development of corticosteroid-induced ocular adverse effects such as cataracts and glaucoma. The vision-robbing consequences of low-grade uveitis occur extremely slowly, typically over a period of 4 to 8 years, and the end result is clear: even low-grade uveitis may lead eventually to ocular damage, including band keratopathy, maculopathy (macular edema, macular cysts, epiretinal membrane), glaucomatous optic neuropathy, and cataract formation from chronic inflammation and corticosteroid therapy. Although surgical treatment of cataract and glaucoma is remarkably successful in the general population, it is consistently less so in patients with JIA-associated uveitis.
The prevalence of JIA in the United States has been estimated recently at 30,000 to 50,000 cases. JIA is more common in girls, with a female-to-male ratio of 3:2. There are 3 types of JIA onset: systemic, which constitutes 11% to 20% of cases; polyarticular (5 or more joints), 17% to 40%; and pauciarticular, 40% to 72%. The peak age at onset of JIA is between 2 and 4 years. The incidence of iridocyclitis in patients with JIA ranges from 8% to 24% and varies among sub-groups of JIA. Uveitis occurs both in children with JIA who are antinuclear antibody (ANA) positive and in those who are ANA negative. Approximately 78% to 90% of patients with JIA-associated iridocyclitis have pauciarticular arthritis; 90% of these patients are ANA positive. Between 7% and 14% of JIA patients with uveitis have polyarticular arthritis; 2% to 6% have systemic arthritis. Uveitis precedes the onset of arthritis in approximately 6% of cases and may be detected at the time of initial diagnosis of arthritis. The majority of patients develop iridocyclitis within 4 to 7 years after JIA onset; the average age at the time of diagnosis of JIA-associated iridocyclitis is 6 to 8 years. The highest risk of iridocyclitis is within 2 years after the onset of arthritis and declines considerably after 8 years have elapsed from the onset of JIA. In some patients, however, the interval between onset of arthritis and uveitis may exceed 20 years. In general, little or no correlation exists between arthritis activity and the presence of uveitis. The uveitis associated with JIA is typically anterior, involving the iris and ciliary body, and generally affects both eyes.
The eyes of patients with JIA-associated uveitis commonly appear normal (not red or inflamed) on external examination and routine ophthalmoscopy. Because patients with JIA are young, they may not notice or report small visual changes that are slowly developing as a result of active inflammation. Therefore, current guidelines recommend that children whose age at onset of JIA is less than 7 years and who do not have known iridocyclitis should have a complete ophthalmologic examination including slitlamp evaluation, every 3 to 4 months if they have pauciarticular or polyarticular JIA and ANA positivity, every 6 months if they have pauciarticular or polyarticular JIA but ANA negativity, and every 12 months if they have systemic JIA. However, Candell, Chalom and colleagues recently reported that, in a retrospective study, although the prevalence of uveitis was lower in ANA-negative patients compared with ANA-positive patients, ocular complications were more common in children with uveitis who were ANA negative. These authors speculated that perhaps ANA-negative children were screened less intensively and thus fared worse. Patients with JIA are considered at low risk for developing iridocyclitis 7 years after the onset of their arthritis but should have yearly ophthalmologic examinations indefinitely thereafter. Once iridocyclitis is diagnosed, the managing ophthalmologist can determine the frequency of visits, depending on the severity of uveitis, its response to therapy, and the treatment used.
A recent retrospective study on predictors of visual outcomes in JIA-associated uveitis reported the characteristics of JIA-associated iridocyclitis in 43 patients with JIA-associated uveitis who were followed for at least 6 months at a tertiary care center. Forty-seven parameters were analyzed to determine the relative odds of visual rehabilitation associated with each characteristic. Thirty-seven patients (86%) were girls. The mean known age of uveitis onset was 13 years; disease onset was 4 years earlier, on average, in girls than in boys. Ninety-three percent of patients with uveitis had chronic inflammation, 5% had recurrent inflammation, and 2% had an acute monophasic disease course. The mean overall duration of uveitis was 146 months, and girls had a significantly longer duration of active disease than boys. After considering all potential confounders, male sex, shorter duration of uveitis, older ages at uveitis onset, and a shorter delay in presentation to a uveitis subspecialist were associated significantly with improvement in visual acuity. Visual acuity at presentation, older age at uveitis onset, use of systemic nonsteroid anti-inflammatory drugs such as methotrexate and/or immunomodulatory drugs, absence of glaucomatous optic neuropathy, and male sex were correlated significantly with a final visual acuity outcome of 20/40 or better.
There are no randomized controlled trails evaluating treatment strategies for JIA-associated uveitis. Observational studies and clinical experience have led us to advocate a treatment philosophy of tolerating no active inflammation at any time in the eyes of children with JIA-associated iridocyclitis. We use a “stepladder” algorithmic approach of progressive aggressiveness of treatment to achieve that goal. The cornerstone of initial management is topical steroid therapy (prednisolone 1%). Regional injection steroids (triamcinolone acetonide) and systemic steroids (prednisone) are often used simultaneously in the initial care of a patient with JIA-associated iridocyclitis. If the inflammation persists after 90 days of treatment and attempted withdrawal of oral and topical steroids, chronic use of an oral NSAID is indicated. Naproxen (available in suspension), 10 to 15 mg/kg per day in 2 divided doses, is a common first choice; tolmetin, 20 to 30 mg/kg per day in 4 divided doses is another choice. These 2 agents have the longest safety record in childhood use. However, pediatricians and pediatric rheumatologists may choose from a wide range of other NSAIDs and dosages for their patients. We usually prescribe a histamine H2-receptor antagonist (ranitidine or famotidine) or the synthetic prostaglandin E1analog misoprostol as prophylaxis for gastritis or ulcerative symptoms for patients who receive NSAIDs, especially if an NSAID is given with a systemic steroid. The Cox-2 specific NSAIDs, such as Celebrex, may become especially appealing for long-term care.
In approximately 30% of cases, JIA-associated iridocyclitis is not satisfactorily controlled by conventional anti-inflammatory treatments, and iridocyclitis recurs whenever steroids are withdrawn despite the longterm use of an oral NSAID. We believe that at this stage advancing to low-dose, once-a-week methotrexate therapy (o.3-o.5 mg/kg per week; typically, 7.5-25 mg per week) should be considered. The dosage of methotrexate can usually be increased to a maximum of 40 mg weekly to control the iridocyclitis if the standard starting dose is not adequate. Therapy with methotrexate, a folic acid antagonist, has had an exceptional safety and efficacy record as used by rheumatologists to care for children with the joint manifestations of JIA. Published data and clinical experience indicate that immunomodulatory agents have considerably fewer adverse effects than systemic corticosteroids when used as single agents in relatively low doses for the treatment of JIA-associated uveitis. The mechanisms of action of methotrexate in JIA are not well understood. However, methotrexate seems to have both antiinflammatory and immunosuppressive actions. The potential for drug-induced adverse effects, such as bone marrow suppression, hepatic toxicity, pneumonitis, and oral ulcerations exists. Clinical studies have shown that the concurrent administration of folic acid or folinic acid with methotrexate does not reduce the efficacy of the drug but might alleviate some of its potential toxicities. The concurrent use of folic acid, 1 mg/d, the close involvement of the physician in longitudinal care, and the appropriate monitoring of hematologic parameters and hepatic enzyme levels minimize the risk of adverse effects. We believe that such careful management accounts for the safety record of low-dose, once-weekly methotrexate. There is no known risk of sterility with low-dose methotrexate therapy. The risk of increased susceptibility to a malignancy later in life also is probably not significant in children with JIA treated with methotrexate. There have been reports of Epstein-Barr virus associated lymphoma and lymphoproliferative disorders occurring in patients with adult RA receiving low-dose methotrexate therapy; discontinuation of methotrexate therapy often leads to complete remission. The authors of these studies acknowledge that methotrexate probably has no direct oncogenic effect itself, but may promote or enhance malignancies associated with Epstein Barr Virus infection in individuals with disorders such as RA. We know of no such predisposition in JIA patients.
When methotrexate treatment fails or is not tolerated, other immunomodulators, such as azathioprine (1-2 mg/kg per day), cyclosporine (2-5 mg/kg per day), mycophenolate mofetil (mg/m2 body surface area bid), or chlorambucil (0.10-0.16 mg/kg per day) may replace methotrexate or be used adjunctively to achieve the goal of total quiescence of ocular inflammation. However, the frequency of failure of methotrexate treatment is quite small. Infliximab or Adalimumab may also be considered.
Further reduction in the occurrence of irreversible blindness secondary to uveitis in patients with JIA depends on early diagnosis of iridocyclitis, facilitated by mandatory vision screening programs in day care centers and schools, and on the use of therapeutic algorithms that include methotrexate and other immunomodulators to eradicate intraocular inflammation. Patients with JIA-associated uveitis should be promptly referred to colleagues experienced with such therapy before permanent ocular damage develops. Our hope is that cooperation among physicians of all specialties caring for patients with JIA will reduce the ocular morbidity and blindness secondary to JIA-associated iridocyclitis.
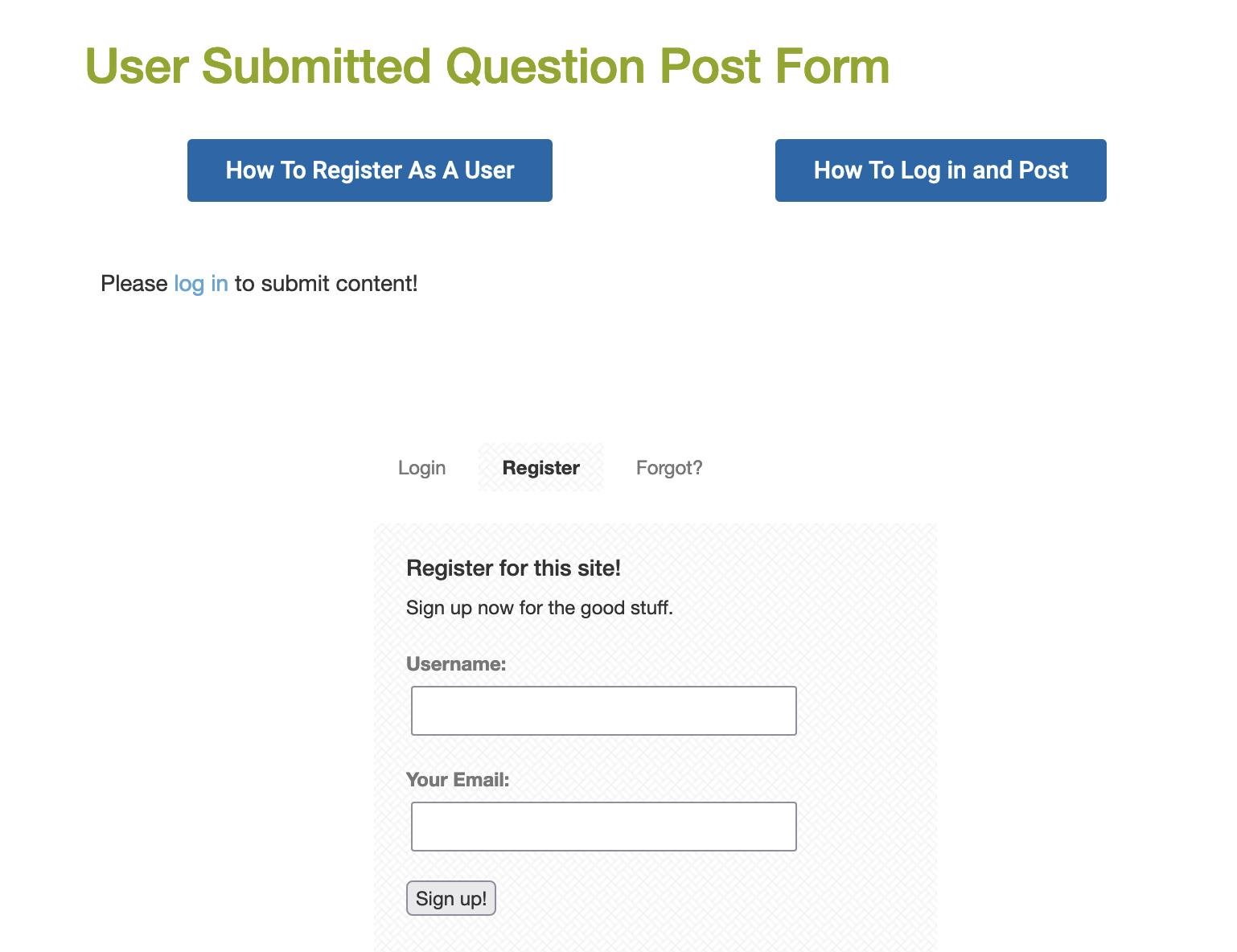
Click on the Register link
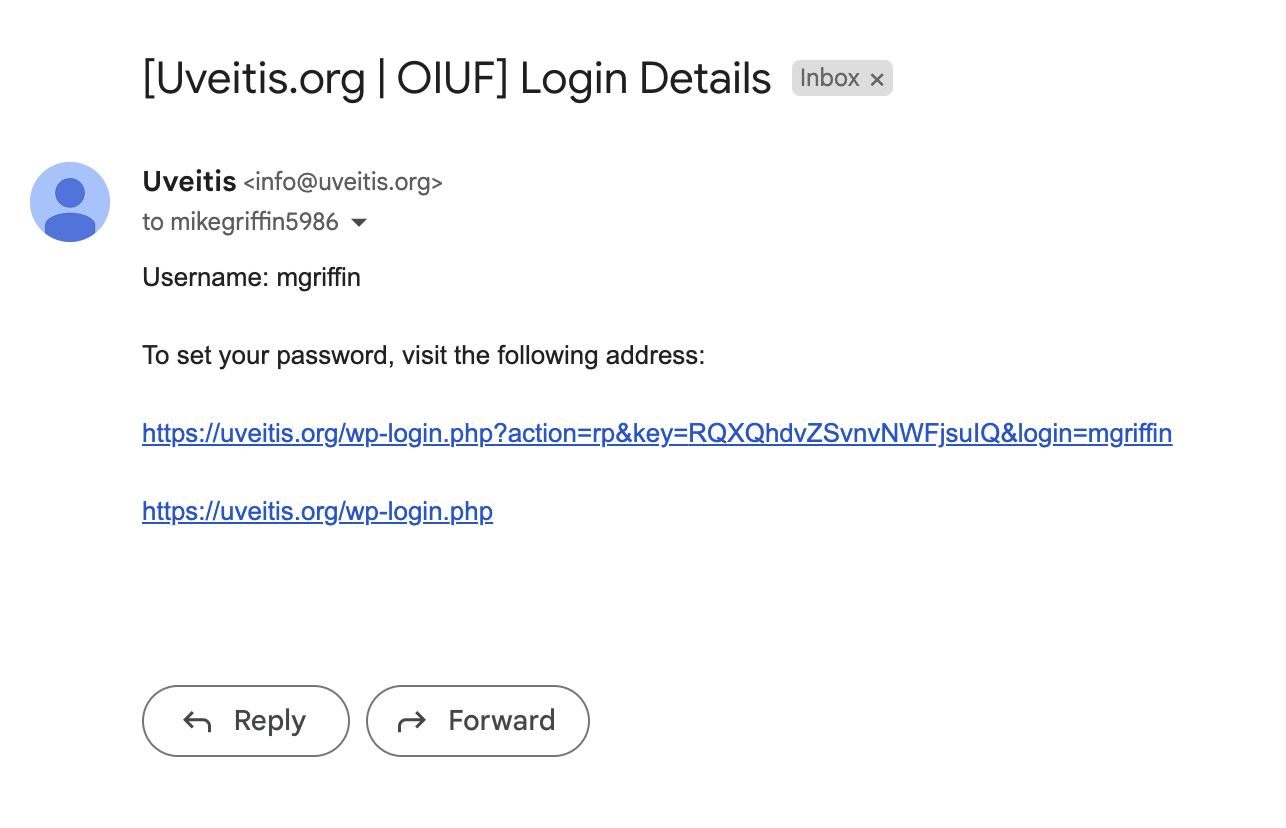
In the email you receive, click on the change password link.
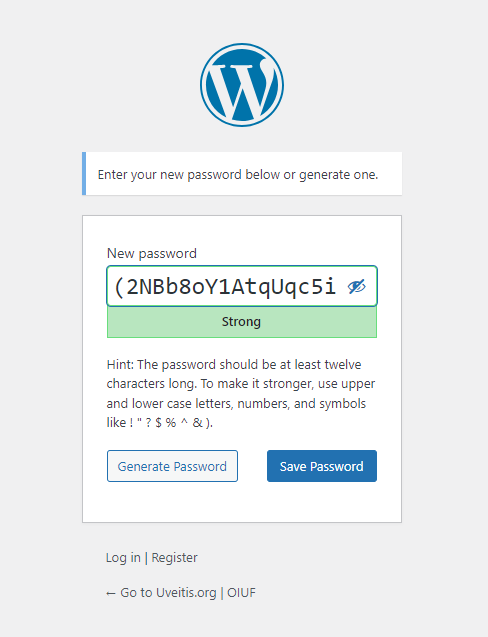
Change your password