Cornea epithelium is subject to constant trauma and shedding of the surface epithelium, and replenishment is from epithelial cells beneath and peripheral to the central desquamating epithelium. The origin of the corneal epithelium appears to reside in the crypts of Vogt, where a population of “immortal” stem cells resides, possessing enormous potential for clonogenic cell division. These cells, like all stem cells, have inherent properties which enable them to accomplish error-free replication which avoids development of abnormal differentiation and cellular dysfunction; a low mitotic rate and asymmetrical DNA segregation are essential in this error-free proliferation of these relatively primitive, poorly differentiated cells. The limbal stem cells lack the keratin 3 differentiation marker typical of all the rest of corneal epithelium, but does possess keratin 19. Cell kinetic studies show that limbal stem cells, and the cells into which they evolve, transiently amplifying cells (TAC) remain in the so called “proliferative” compartment, undergoing cell division and not evolving to commitment to further specialized differentiation. These cells (stem cells and TAC) are located exclusively in the limbal region, with the stem cells in the basal epithelium and TAC occurring in the basal and suprabasal levels, extending up to the superficial layers. The corneal epithelium is then maintained by cellular proliferation of these cells which then migrate further onto the central cornea and become terminally differentiated. The lack of a vascular supply and the presence of special elements within the corneal basement membrane, and differences in vitamin A concentrations may be some of the primary factors responsible for the terminal differentiation events.
The absence or malfunction of corneal stem cells is characterized by the loss of proliferative capacity of corneal epithelium, resulting in surfacing of the cornea with “transdifferentiated” conjunctivally-derived epithelium or, in the worst case, failure to resurface at all in the presence of a persistent epithelial defect, with corneal neovascularization and scarring. Such disorders include primary dysfunctions such as aniridia and congenital erythrokeratoderma, and secondary ones (the most common, in which limbal stem cells are destroyed, either traumatically (e.g., alkali burns) or immunologically (e.g., Stevens Johnson syndrome). It is well recognized that simply replacing the cornea through corneal transplantation in these circumstances is almost uniformly unsuccessful. Limbal stem cell grafting has changed all that, but we should emphasize here that, unless the underlying primary problem has been corrected (for example, the constant assault onto the ocular surface from external aggravants, such as sicca syndrome, meibomian gland dysfunction, lagophthalmos, trichiasis, distichiasis, and the “sandpapering” effect of the keratinizated posterior lid margin), limbal stem cell grafting will not be the panacea for such disorders either. However, if one can correct those underlying saboteurs, then limbal stem cell grafting may set the stage for the ultimate visual rehabilitation step (i.e., keratoplasty), once the ocular surface has been re-established with normal, corneally-derived epithelium.
Our experience with 19 patients who have undergone limbal stem cell grafting has been quite gratifying. All have been patients with rather desperate situations, most commonly in the context of Stevens-Johnson syndrome, but in some instances in the context of cicatricial pemphigoid or severe atopy. We have had two failures as a result of limbal stem cell graft rejection, and three secondary to infection. All the patients are at high risk for infection because of the debilitated ocular surface in the first place, coupled with the fact that all are on topical steroid. The use of prophylactic topical antibiotic and of amniotic membrane grafting in order to protect the grafted limbal stem cells appears to be reducing the infection rate, but of course only time will tell on this matter.
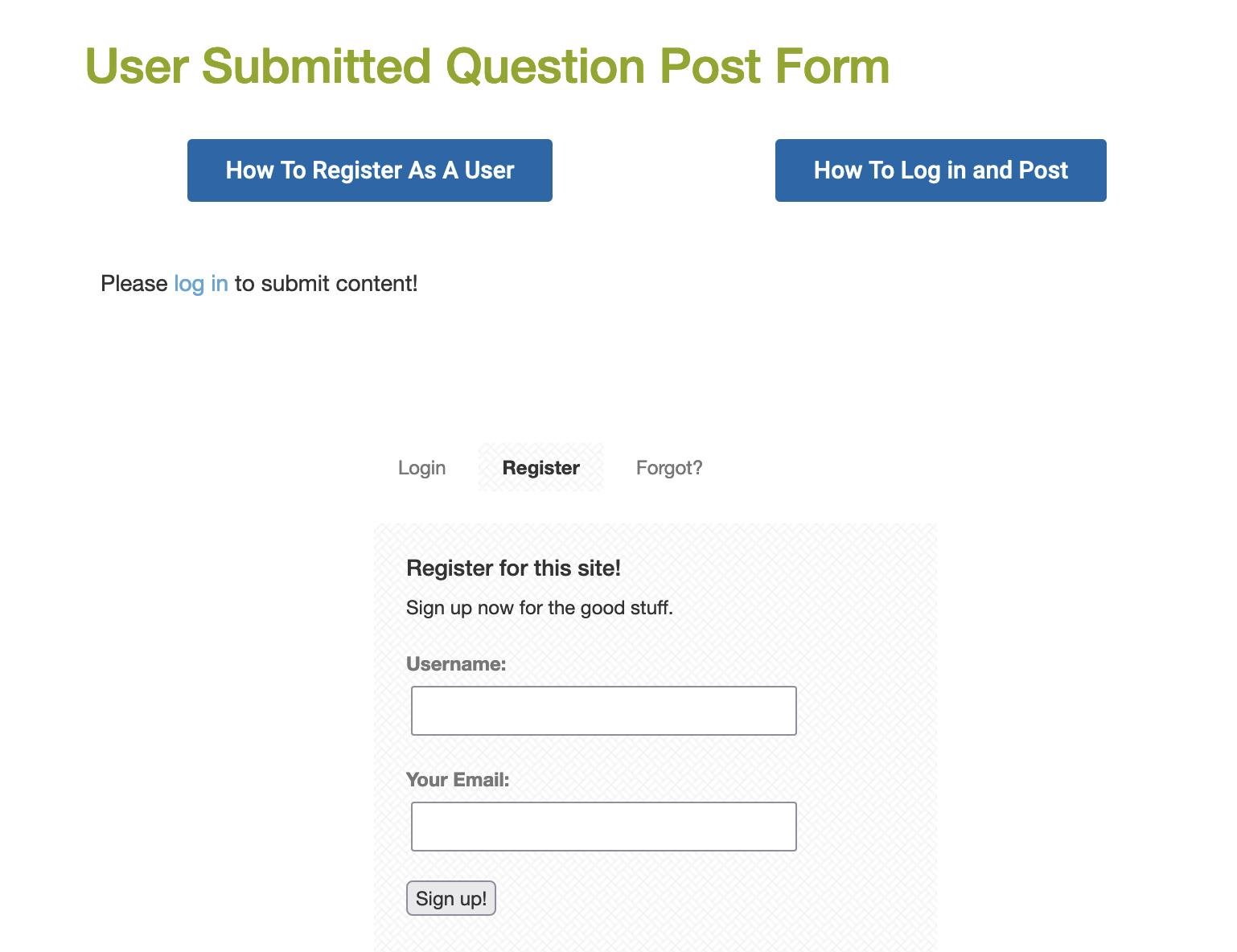
Click on the Register link
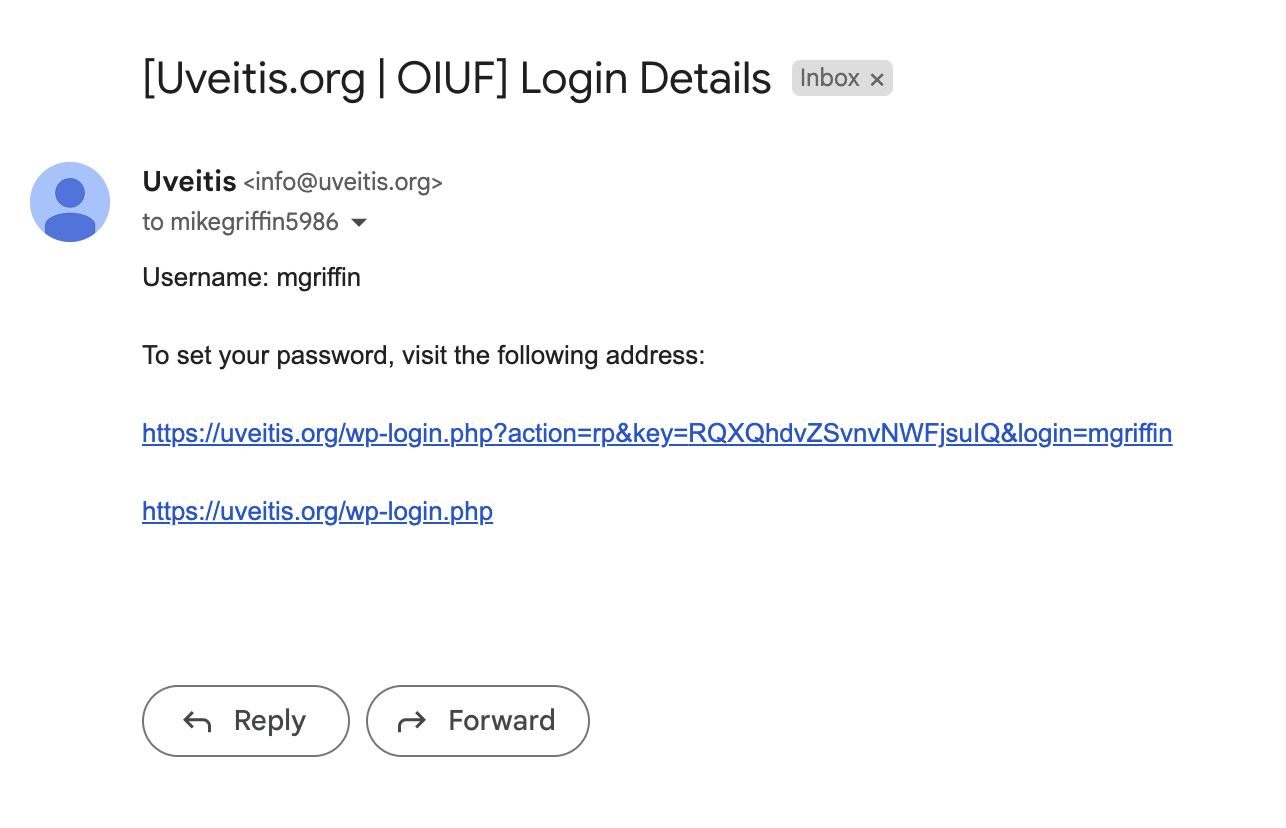
In the email you receive, click on the change password link.
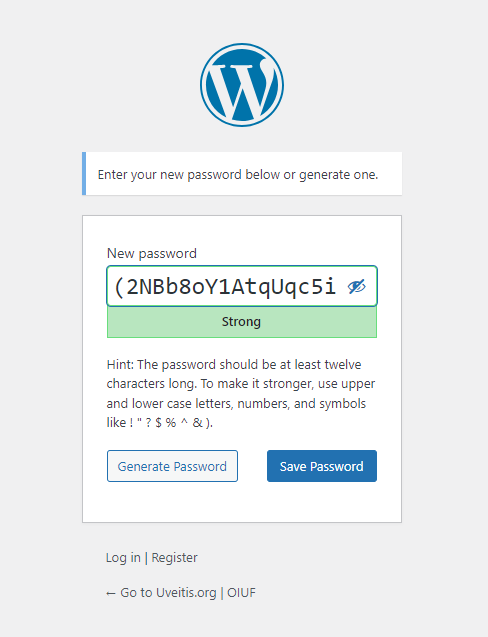
Change your password